
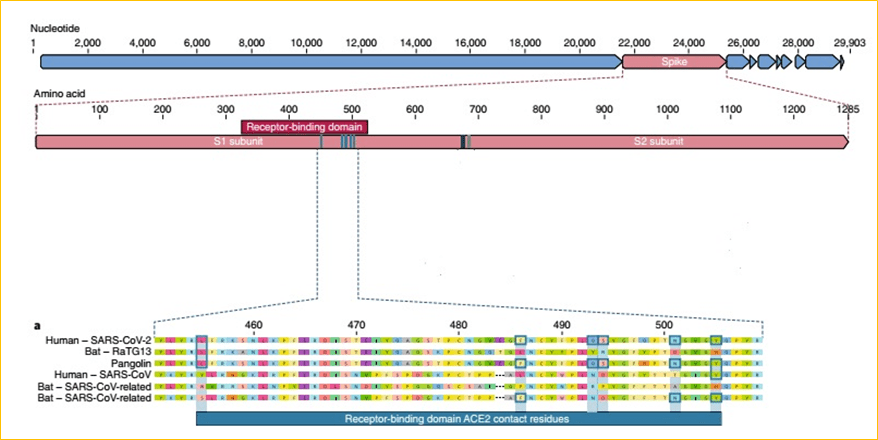
1. Mutations in the receptor-binding domain of SARS-CoV-2:
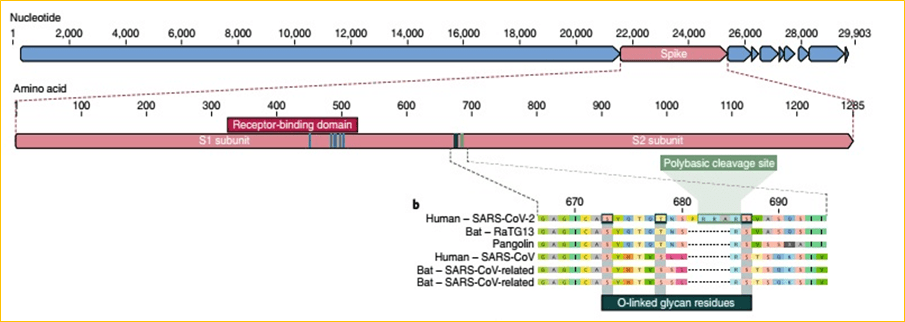
The receptor binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-2. Five of these six residues differ between SARS-CoV-2 and SARS-CoV. The high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise.
2. Polybasic furin cleavage site and O-linked glycans
A unique polybasic cleavage site (RRAR) at the junction of S1 and S2, the two subunits of the spike. This allows effective cleavage by furin and other proteases and has a role in determining viral infectivity and host range. In addition, a leading proline is also inserted at this site in SARS-CoV-2; thus, the inserted sequence is PRRA. This results in the addition of O-linked glycans to S673, T678 and S686. The function of the predicted O-linked glycans is unclear, but they could create a‘mucin-like domain’ that shields epitopes or key residues on the SARS-CoV-2 spike protein.
It is improbable that SARS-CoV-2 emerged through laboratory manipulation of a related SARS-CoV-like coronavirus. As noted, the RBD of SARS-CoV-2 is optimized for binding to human ACE2 with an efficient solution. Furthermore, if genetic manipulation had been performed, one of the several reverse-genetic systems available for betacoronaviruses would probably have been used. However, the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used virus backbone. Instead, there are two scenarios that can plausibly explain the origin of SARS-CoV-2:
(i) natural selection in an animal host before zoonotic transfer; and
(ii) natural selection in humans following zoonotic transfer.
As many early cases of COVID-19 were linked to the Huanan market in Wuhan, it is possible that an animal source was present at this location. Similarity of SARS-CoV-2 to bat SARS-CoV-like coronaviruses, bats serve as reservoir. Pangolin coronaviruses exhibit strong similarity to SARS-CoV-2 in the RBD, including all six key RBD residues. But both bat and pangolin lacks polybasic cleavage site. Mutations, insertions and deletions can occur near the S1–S2 junction of coronaviruses, which shows that the polybasic cleavage site can arise by a natural evolutionary process. For a precursor virus to acquire both the polybasic cleavage site and mutations in the spike protein suitable for binding to human ACE2, an animal host would probably have to have a high population density (to allow natural selection to proceed efficiently) and an ACE2-encoding gene that is similar to the human ortholog.
It is possible that a progenitor of SARS-CoV-2 jumped into humans, acquiring the genomic features described above through adaptation during undetected human-to-human transmission. Once acquired, these adaptations would enable the pandemic to take off and produce a sufficiently large cluster of cases to trigger the surveillance system that detected it. The presence of an RBD in pangolins very similar to that of SARS-CoV-2 means that we can infer this was also probably the virus that jumped to humans, but no polybasic site. Emergence of the virus in late November 2019 to early December 2019, compatible with the earliest retrospectively confirmed cases. Hence, this scenario presumes a period of unrecognized transmission in humans between the initial zoonotic event and the acquisition of the polybasic cleavage site. Not leaked from cell-culture labs, O-linked glycans can occur only in presence of an immune system.
Source: Nature
